Een ernstige erfelijke aandoening is nu te behandelen met een minuscuul chirurgisch sneetje – niet in het weefsel, maar in het dna van stamcellen van de patiënt. Als eerste ter wereld gaf de Britse medicijnautoriteit MHRA op 15 november voorwaardelijke goedkeuring aan een genetische behandeling op basis van het moleculaire knipgereedschap crispr-cas9. De behandeling is gericht tegen twee ernstige bloedziekten: sikkelcelziekte en bèta-thalassemie. Het is de allereerste klinische toepassing van het revolutionaire genetische knip-en-plakgereedschap crispr-cas9.
„Dit kwam verbluffend snel”, zegt Sjaak Philipsen, hoogleraar moleculaire celbiologie aan het Erasmus MC in Rotterdam. Crispr-cas9 werd in 2012 ontdekt en in 2020 bekroond met een Nobelprijs. Met de techniek kunnen genen heel precies worden aan- of uitgezet en bewerkt.
De techniek is een welkome toevoeging aan het arsenaal van mogelijkheden om genetische aandoeningen te behandelen. De schaarse gentherapieën tot nu toe maken gebruik van onschuldige virussen om correcte genen in cellen te brengen.
Lees ook
Eerste baby in Nederland behandeld met gentherapie van eigen stamcellen
ontwikkeld door het Amerikaanse bedrijf Vertex Pharmaceuticals en het Zwitserse biotechbedrijf CRISPR Therapeutics.
„Fenomenaal dat dit is goedgekeurd”, reageert Niels Geijsen, hoogleraar regeneratieve geneeskunde aan het LUMC in Leiden. „Het is een teken dat we een nieuw tijdperk ingaan waarin we mogelijk oplossingen kunnen vinden voor ziekten die voorheen onbehandelbaar waren.” Vier vragen over het geavanceerde genetische medicijn.
Lees ook
CRISPR-Cas ontketent een tweede genetische revolutie
1Tegen welke ziekten werkt deze crispr-cas9-behandeling?
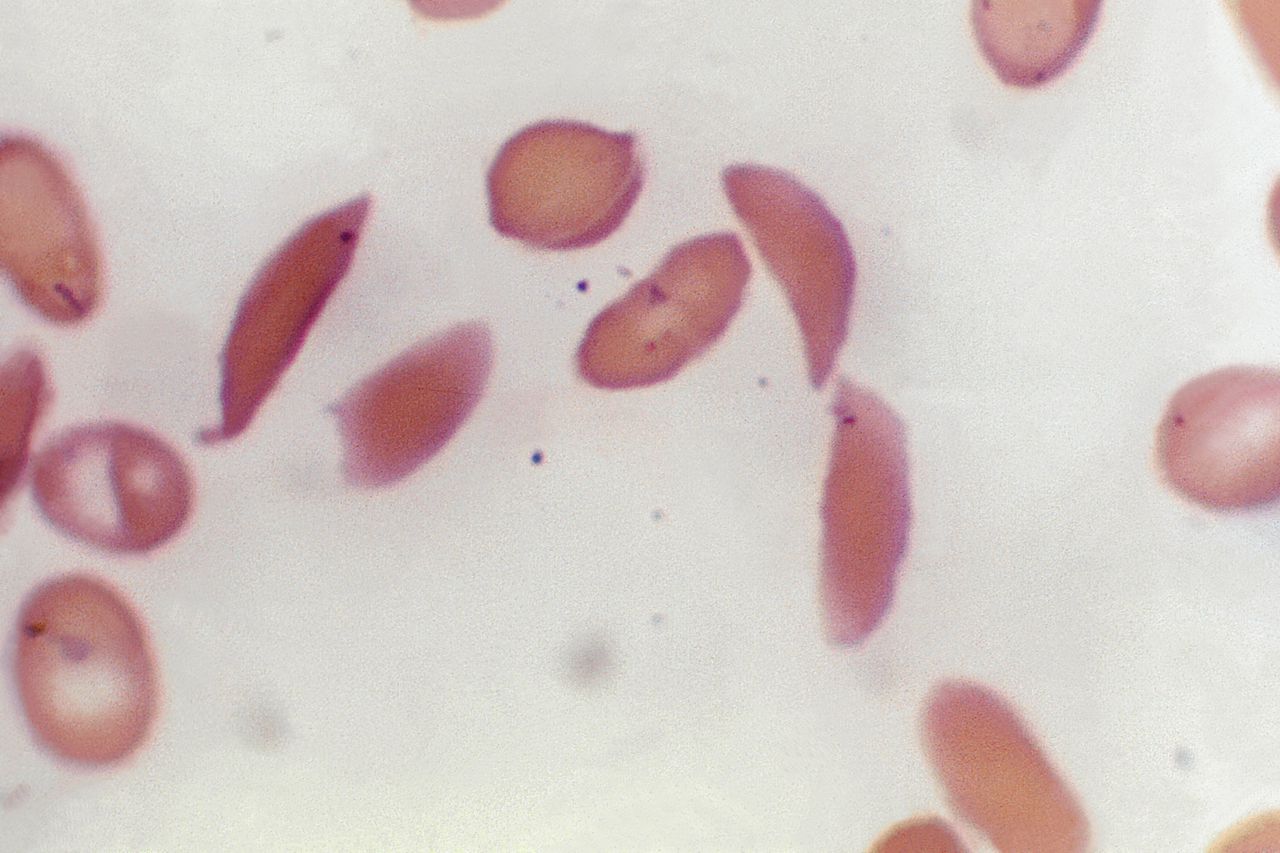
Sikkelcelziekte en bèta-thalassemie zijn erfelijke vormen van bloedarmoede. Ze worden veroorzaakt door fouten in de genetische code voor hemoglobine, een cruciaal eiwit in rode bloedcellen dat zuurstof bindt. Dankzij dat eiwit kunnen rode bloedcellen alle uithoeken van het lichaam van zuurstof voorzien. Hemoglobine bestaat uit vier bouwstenen: twee alfa-globine en twee bèta-globine. Bij de genoemde aandoeningen zitten de defecten in het bèta-globine.
Bij mensen met sikkelcelziekte, ook wel sikkelcelanemie, worden door de mutatie de rode bloedcellen sikkelvormig. De cellen worden binnen een maand afgebroken (gezonde rode bloedcellen pas na 120 dagen), wat tot bloedarmoede leidt. Patiënten voelen zich vaak moe, kortademig en duizelig. Daarnaast kunnen de sikkelcellen kleine bloedvaatjes verstoppen. „Dat veroorzaakt plotselinge hevige pijn-aanvallen, doordat botten of organen lokaal niet genoeg zuurstof krijgen”, zegt Philipsen. Zijn groep werkt onder meer aan genetische behandelingen tegen deze sikkelcelziekte. „In de loop van het leven ontstaat steeds meer orgaanschade. De ziekte kan ook leiden tot levensbedreigende infecties, geelzucht of galstenen. In Nederland is de levensverwachting van deze patiënten gemiddeld 25 jaar minder dan van gezonde mensen, en er overlijden ook kinderen en tieners aan.” Bij bèta-thalassemie wordt te weinig bèta-globine gemaakt, wat tot een tekort aan rode bloedcellen leidt.
Sikkelcelziekte komt veel voor in India en in Afrikaanse landen ten zuiden van de Sahara, bèta-thalassemie komt het meest voor in landen rond de Middellandse Zee, het Midden-Oosten en in delen van Azië en Afrika. „De mutaties beschermen de dragers een beetje tegen malaria. Daardoor zijn er veel patiënten in landen waar malaria heerst of heerste”, zegt Philipsen. „Er zijn zeven miljoen mensen met sikkelcelziekte in de wereld. Bijvoorbeeld in Nigeria worden er 150.000 kinderen per jaar mee geboren.” Bèta-thalassemie komt minder vaak voor. In Nederland hebben ongeveer 2.000 mensen sikkelcelziekte, en zo’n 200 mensen bèta-thalassemie.
De behandeling van bèta-thalassemie bestaat uit maandelijkse bloedtransfusies om rode bloedcellen aan te vullen. Bij sikkelcelziekte hangt de behandeling af van de symptomen van de patiënt, in het arsenaal zitten onder meer bloedtransfusies, antibiotica en pijnstillers. Bij een ernstig verloop kan een stamceltransplantatie nodig zijn. Daarbij worden de bloedvormende stamcellen in het beenmerg vervangen door die van een gezonde donor. Het is een zware ingreep die alleen kan worden gedaan als er een passende donor beschikbaar is, en waarbij er risico is op afstoting. Dat risico is er niet als er gebruik wordt gemaakt van de eigen stamcellen, zoals bij de nieuwe therapie.
2Hoe werkt de crispr-cas-gentherapie tegen deze bloedziekten?
Met de behandeling wordt in rode bloedcellen een zuurstofbindend eiwit geactiveerd dat normaal alleen bij foetussen en pasgeboren baby’s actief is: gamma-globine. Zo wordt via een omweg het hemoglobine in rode bloedcellen hersteld, zodat die weer zuurstof kunnen rondbrengen. Bij deze behandeling ontvangt de patiënt geen stamcellen van een donor, maar krijgt de eigen, in het laboratorium aangepaste stamcellen weer terug.
In een kweekschaal wordt crispr-cas9 bij de stamcellen van de patiënt gevoegd. Dat genetische gereedschap bestaat uit een specifiek rna-molecuul en een enzym dat dna kan knippen, cas9. Het rna-molecuul leidt cas9 naar het BCL11A-gen. Dat wordt geknipt, zodat het niet meer actief is.
BCL11A wordt bij mensen na de geboorte actief in rode bloedcellen. Het voorkomt dat gamma-globine wordt gemaakt. Door de knip in het BCL11A-gen wordt in de rode bloedcellen die uit de stamcellen ontstaan weer het foetale hemoglobine gemaakt. Zo kunnen die rode bloedcellen hun normale vorm houden en zuurstof rondbrengen.
Voordat de aangepaste stamcellen via een infuus weer het lichaam in worden gebracht, moeten in het beenmerg de resterende eigen stamcellen worden gedood met chemotherapie. Patiënten moeten zeker een maand in het ziekenhuis blijven voordat de nieuwe cellen in het beenmerg zijn beland en voldoende nieuwe bloedcellen maken.
3Hoe veilig is de genetische therapie?
Aan het onderzoek op basis waarvan de Britse medicijnautoriteit de behandeling voorwaardelijk goedkeurde, deden 45 mensen met sikkelcelziekte mee. Daarvan konden 29 lang genoeg gevolgd worden om tussentijdse conclusies te kunnen trekken. Van die mensen bleven er 28 minstens een jaar lang verlost van de pijnlijke sikkel-crises, schrijft de MHRA in hun persbericht.
In een tweede studie kregen 54 mensen met een ernstige vorm van bèta-thalassemie de behandeling. Daarvan deden 42 lang genoeg mee om het effect te beoordelen. Van hen hadden 39 patiënten het hele jaar erop geen bloedtransfusies nodig. De overige drie hadden er 70 procent minder nodig. „Dat geeft deze mensen een enorme vrijheid”, zegt Geijsen.
Een stamceltransplantatie brengt risico’s mee. De bijwerkingen die optraden bij de patiënten in de studies hadden te maken met die ingreep, en waren onder meer misselijkheid, vermoeidheid, koorts en een verhoogd risico op infecties. Beide studies lopen door en worden gevolgd door de fabrikant en door de medicijnautoriteit. De voorwaardelijke goedkeuring geldt een jaar, daarna worden de nieuwste resultaten meegenomen bij een nieuwe beoordeling.
Leven met gamma-globine in de rode bloedcellen is geen probleem, zegt Philipsen. „En zijn families bekend bij wie gamma-globine ook nog in het bloed aanwezig is. In het dagelijks leven heb je daar geen last van. Alleen bij topsport of op grote hoogte is het misschien niet optimaal, omdat deze vorm van hemoglobine zuurstof iets minder makkelijk afstaat.”
Dat de eerste ziekten waarbij crispr-cas9 wordt ingezet bloedziekten zijn, is geen toeval. „Beenmerg kun je uit het lichaam nemen om de stamcellen in het lab te bewerken”, zegt Geijsen. „Het genetische gereedschap hoeft niet het lichaam in. Behandelingen met crispr-cas9 die wel het lichaam in gaan worden ook al uitgetest. Die richten zich op organen waarbij de bewerking lokaal blijft, zoals bijvoorbeeld het oog.”
Nóg geavanceerdere crispr-technologie kan gericht een mutatie in een gen repareren, bijvoorbeeld in het bèta-globinegen zelf. „Maar dat inzetten als behandeling is nog een stuk verder weg. Die technieken werken soms minder efficiënt of hebben nog technische beperkingen”, zegt Geijsen. „Maar het veld ontwikkelt zich snel, ik verwacht dat er de komende jaren veel nieuwe mogelijkheden ontstaan.”
4Is de behandeling de redding voor alle mensen met sikkelcelziekte?
Dat is onwaarschijnlijk. In de eerste plaats zal de behandeling erg duur zijn, naar schatting 2 miljoen euro per patiënt. Het Zorginstituut Nederland verwacht er tien tot dertig patiënten per jaar mee te behandelen, mocht de behandeling worden goedgekeurd.
In Nederland onderzoeken de bloedbank Sanquin, het Hubrecht Instituut in Utrecht en de universitaire medische centra van Amsterdam, Leiden en Rotterdam of het haalbaar is om zelf een vergelijkbare behandeling te ontwikkelen. Philipsen: „Hopelijk helpt dat om de prijs naar beneden te krijgen zodat het breder toegankelijk wordt. Echt goedkoop wordt het nooit, maar de helft van deze prijs zou al een stuk beter zijn.”
Maar zelfs als het goedkoper wordt, zullen de meeste patiënten er niet mee geholpen kunnen worden. „Veruit de meeste mensen met deze ziekten wonen in landen die de infrastructuur niet hebben om deze hoog-technologische behandeling te kunnen bieden en de patiënten te blijven volgen”, zegt Geijsen.
Toch ziet Geijsen grote voordelen. „Mijn hoop is dat dankzij deze grote stap de techniek meer geaccepteerd raakt. We zullen ontzettend veel leren van deze eerste stap, waardoor we ook toepassingen in andere behandelingen zullen kunnen ontwikkelen. Zo is de verwachting dat deze behandeling iemand levenslang geneest, maar dat moet natuurlijk nog blijken.”
Vooralsnog is de behandeling nu in het VK voorwaardelijk goedgekeurd. In de Verenigde Staten buigt de voedsel- en medicijnenautoriteit (FDA) zich over de aanvraag, na een positief advies van een externe wetenschappelijke commissie op 31 oktober. Ook bij het Europese medicijnagentschap EMA loopt de beoordeling. Mocht hun oordeel positief uitvallen, dan zal naar verwachting rond februari 2024 registratie volgen.